Q. Cui and M. Karplus
Dept. of Chem. & Chem. Biol.
Harvard University
The main theme of our research is to understand the dynamics of biomolecules and enzymatic catalysis using theoretical approaches. For this purpose, we use the combined quantum mechanical and molecular mechanical (QM/MM) method, which has been under continuous development over the past decade or so in the current research group. [1] Using the parallel computational resources at the Argonne National Laboratory, we have studied a number of systems including the triosephosphate isomerase (TIM), and myoglobin-CO/O2.
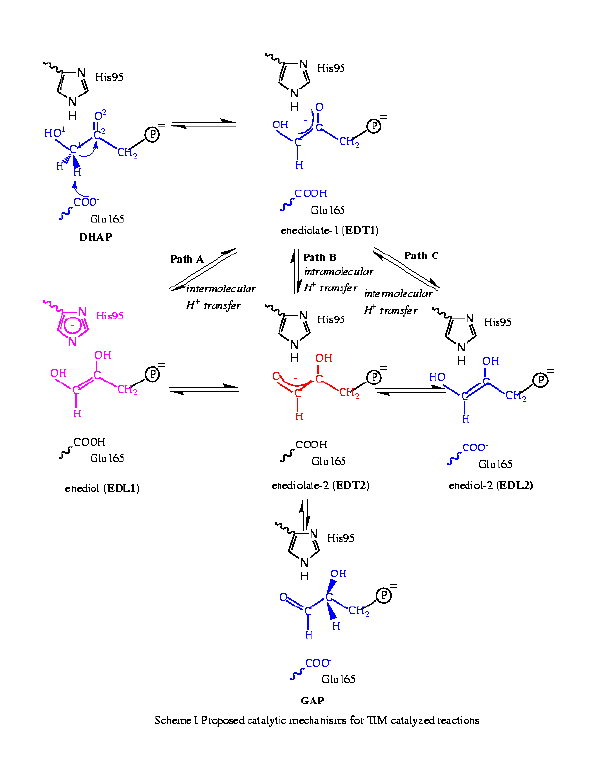
The triose phosphate isomerase (TIM) is a dimeric enzyme that catalyzes the conversion between dihydroxyacetone phosphate(DHAP) and R-glyceraldehyde 3-phosphate (GAP), which is an important step in the glycolytic pathway. Despite numerous experimental [2] and theoretical [3] studies, the precise catalytic mechanism for the TIM reactions remains to be clarified. Three paths, A, B and C, have been proposed, which are shown in Scheme I.
The energetics associated with these paths were studied using the QM/MM approach at three QM levels including AM1, AM1-SRP (Specific Reaction Parameters) and B3LYP/6-31+G(d,p). Selected active site residues are shown in Figure 1, and the schematic potential energy profiles are shown in Figure 2.
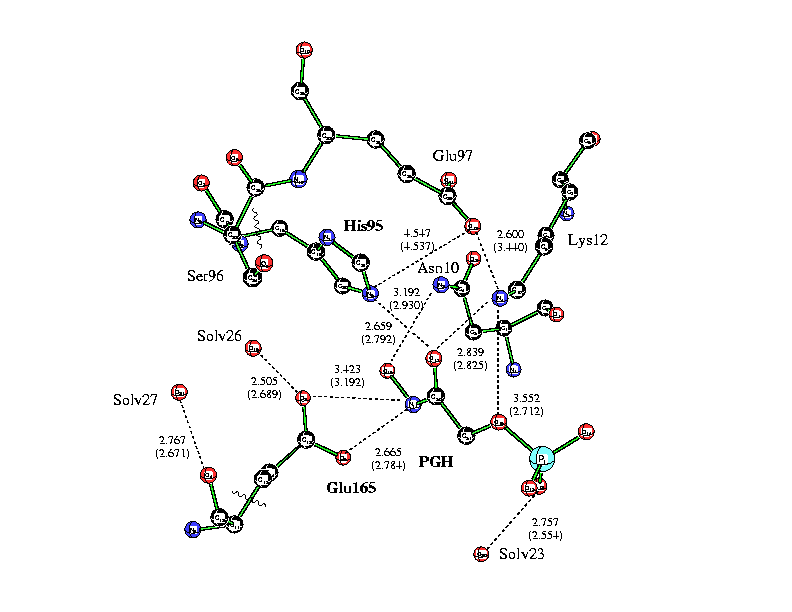
Figure 1
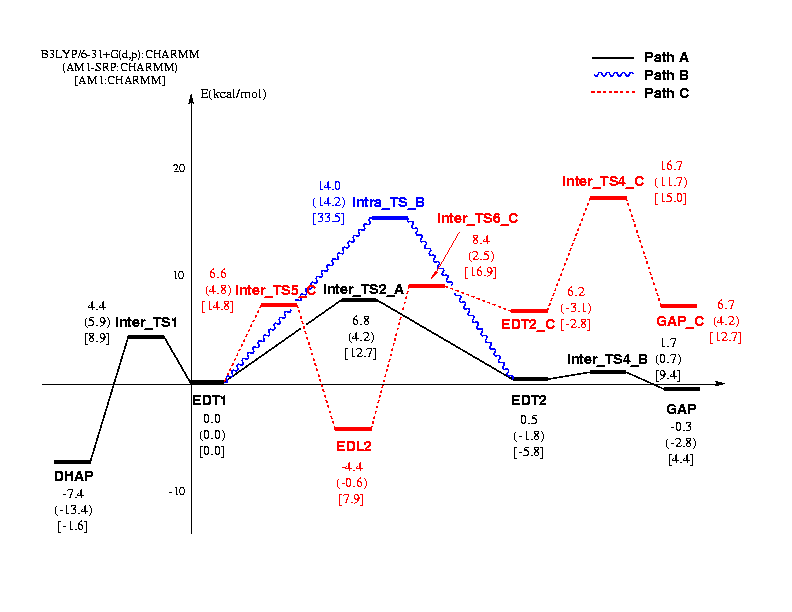
Figure 2
The path A involves His95 as the general acid and has been found to be energetically most favorable. The rate-limiting step is the proton abstraction from the substrate Ca by Glu165, which has a barrier of 11.8kcal/mol, in reasonable agreement with the experimental estimate of 13.0kcal/mol. The highest barrier in path C, which was originally proposed for H95Q mutant, is found to be only 1.0kcal/mol higher, and corresponds to the conversion from EDL2 to EDT2_C. Therefore path C may also contribute in reality, which has also been proposed by Mildvan et al. based on NMR and isotope labeling experiments [4]. The path B, which involves an intramolecular proton transfer in the enediolate species, was found to have an even higher barrier of 14.0kcal/mol due to the presence of the His95 in the active site. Through perturbational analyses, it was found that Lys12 and a few polar residues such as Asn10 and Ser211 plus a number of water molecules in the active site have the largest contributions. Nevertheless, a few residues that are reasonably far from the active site such as Gly210 also make notable contributions. The fact that active site water molecules make important contributions can be related to a number of mutation studies from Petsko and co-workers [5].
Another interesting issue related to the TIM system is the importance of proton tunneling. Kinetic isotope effect measured by Klinman et al. [6] for a number of enzymes including alcohol dehydrogenase, amine oxidases and lipoxygenase suggested that proton tunneling is non-negligible even at room temperature. However, detailed understanding of the tunneling effect in enzymes is still lacking. For instance, one interesting question is whether tunneling is more important in enzyme than in solution such that it actually plays a role in catalysis. We used the VTST-SAG (variational transition state theory with semiclassical ground state multidimensional tunneling) approach to determine the magnitude of tunneling in two proton transfer steps catalyzed by TIM. The results are also compared to that for the corresponding reactions in the gas phase and in solution. The results obtained so far suggest that tunneling is less significant in enzyme than in the gas phase, and even less so in solution. More calculations with different environmental configurations of the active site are being carried out.
Another protein studied is the liganded (CO/O2)
myoglobin. The binding differentiation of the two ligands towards myoglobin
has been a long-standing issue in biochemistry, and has been studied by
numerous experimental and theoretical work. With the QM/MM approach, the
issue can be examined at a new resolution. The structures of liganded myoglobin
were optimized with the porphine ring, the ligand and the proximal histidine
treated by B3LYP associated with double-zeta quality basis set. The QM
model was also studied in the gas phase to investigate the effect of the
protein environment. It was found that the protein does have a significant
effect on the relative binding energies of CO and O2, which
is about 20.5kcal/mol in favor of CO in the gas phase and reduced to about
7.0kcal/mol in the enzyme. The electrostatic contribution of each residue
was obtained through a perturbational analysis, and the results are presented
in Figure 3.
Figure 3
Evidently, the distal histidine makes the largest contribution
of ~2.6kcal/mol in favor of O2 binding, in accord with the more
favorable hydrogen-bonding interaction with them in Mb·O2.
A few other residues such as R45, K63 also make sizable contributions.
These results can be related to the recent mutation studies of Phillips
and co-workers [7]. Also being calculated is the normal modes of Mb·CO,
which is related to the recent polarized IR measurements on the CO orientation [8].
Such a calculation is made possible by the recent implementation of analytical
QM/MM Hessian [9].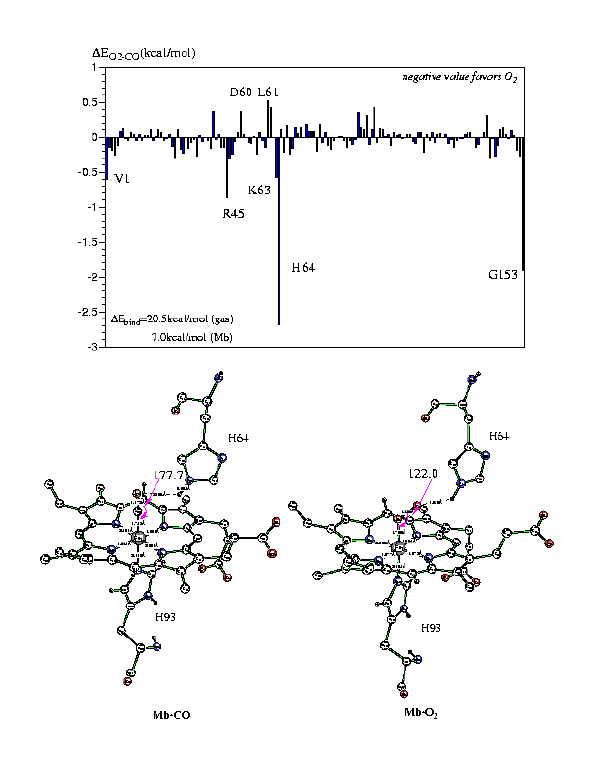
A number of other systems are currently under study with the new QM/MM methods that have been developed recently in this group. An implementation of the algorithm to calculate NMR chemical shielding tensors in the QM/MM framework makes it possible to study the chemical shift of specific group in biomolecules [10]. Combining such a method with molecule dynamics allows one to determine the contribution of the chemical shielding anisotropy to spin relaxation. Currently, calculations are being carried out for a t-RNA and a small lead-dependent ribozyme. In another study, a semi-empirical DFT method, SCC-DFTB (Self-Consistent-Charge Density-Functional Tight Binding) was implemented into CHARMM [11]. It was shown that this method is in general more accurate than AM1 and PM3 at similar computational cost, and therefore can be directly applied as the QM level in QM/MM calculations to obtain semi-quantitative information about macromolecules. Systems currently understudy include the horse liver alcohol dehydrogenase and bacterial photosynthetic center. Other developments in QM/MM methodology include multi-layer methods and implicit solvation models [12].
In conclusion, the parallel computational facility at Argonne has been crucial to our studies of several interesting biomolecules, and the developments of new computational methods. We hope that such support will continue in the future.
References
1. (a) P. A. Bash, M. J. Field, and M. Karplus, J. Am. Chem. Soc. 109, 8092 (1987); (b) M. J. Field, P. A. Bash, and M. Karplus, J. Comp. Chem. 11, 700 (1990)
2. For a recent review, see J. R. Knowles, Nature, 350, 121 (1991)
3. (a) P. A. Bash, M. J. Field, R. C. Davenport, G. A. Petsko, D. Ringe, an d M. Karplus, Biochem. 30, 5826 (1991); (b) G. Alagona, C. Ghio, P. A. Kollman, J. Am. Chem. So c. 117, 9855 (1995); (c) J. \201qvist and M. Fothergill, J. Bol. Chem. 271, 10010 (1996)
4. (a) T. K. Harris, C. Abeygunawardana, and A. S. Mildvan, Biochem. 36, 14 661 (1997); (b) T. K. Harris, R. N. Cole, F. I. Comer and A. S. Mildvan, Biochem. 37, 16828 (1998)
5. ZD, Zhang, E. A. Komives, S. Sugio, S. C. Blacklow, N. Narayana, N. H. X uong, A. M. Stock, G. A. Petsko and D. Ringe, Biochem. 38, 4389 (1999)
6. For a recent review, see, A. Kohen and J. P. Klinman, Acc. Chem. Res. 31 , 397 (1998)
7. (a) J. S. Olson, G. N. Phillips, Jr. J. Biol. Inorg. Chem. 2, 54 4 (1997); (b) G. N. Phillips, Jr., M. L. Teodoro, T. Li, B. Smith, and J. S. Olson, J. Phys. Chem. B 103, 8817 (1999)
8. M. Lim, T. A. Jackson and P. Anfinrud, Science, 269, 962 (1995)
9. Q. Cui, M. Karplus, J. Chem. Phys. in press.
10. Q. Cui, M. Karplus, to be submitted
11. Q. Cui, M. Elstner et al. in preparation.
12. Q. Cui, M. Karplus, in preparation.